LigandMPNN for Inverse Folding 🔙
Inverse Protein Folding is the step following protein backbone generation in protein design pipelines. It involves designing amino acid sequences that will fold into a given protein backbone structure. ProteinMPNN tackles this problem by using a message-passing neural network architecture to design sequences conditioned on backbone structure.
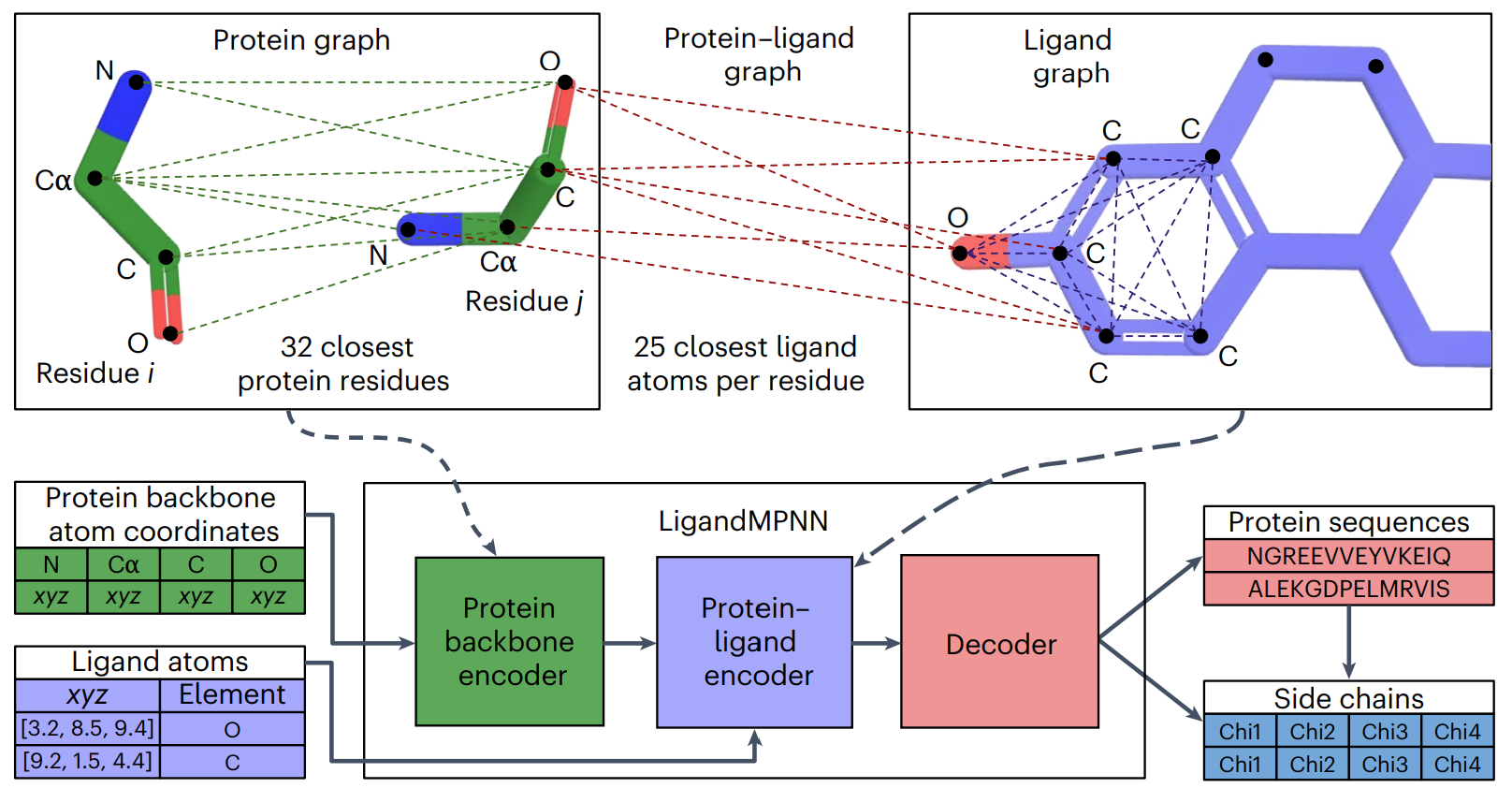
LigandMPNN extends this method to include ligand information, thus conditioning the designed sequence on the atomic context as well. It shows increased recovery of native sequences in pockets interacting with small molecules, nucleotides and metals.
This space allows you to run inverse folding jobs using Hugging Face's hardware and download the results! It is based on LigandMPNN's original Github repository.
Image and Model Source: Dauparas, J., Lee, G.R., Pecoraro, R. et al. Atomic context-conditioned protein sequence design using LigandMPNN. Nat Methods 22, 717–723 (2025). https://doi.org/10.1038/s41592-025-02626-1
{kind=link}
How to Use this Space
Refer to the original repo for a detailed description of the available command line arguments. While essential parameters
such as number of designs to generate, temperature, and which chains to design can be easily controlled through the UI, more advanced parameters can still be specified under Advanced Options.
Please note that this space hard-codes the version of LigandMPNN weights so that trying to change the checkpoint using --checkpoint_ligand_mpnn will cause errors.
Batch generation allows to design sequences for multiple PDB files at once. Note that CLI options --fixed_residues_multi, --redesigned_residues_multi and the like that allow fine-grained
pdb-specific control over design parameters are not yet implemented. However, one can still specify which residues to fix for all PDBs in the batch at once using --fixed_residues.
This space pairs well with the RFD3 backbone generation space as the PDB files in its output can be directly uploaded for batch generation.